
胰腺癌中有80%以上的患者是胰导管癌(PDAC),PDAC是一种高度侵袭性的消化系统肿瘤,起源于胰腺导管上皮及腺泡细胞,是人类最为致命的恶性肿瘤,被医学界称为“癌中之王”。即便近十几年来在癌症治疗上有了巨大的进步,PDAC的五年生存率依然很低。当前对PDAC的发病机制尚不明确。因此,对 PDAC的生长和转移机制研究、寻找潜在的治疗靶点和新的生物标志物是十分必要的。在以往的研究中发现,很多与代谢相关的基因和PDAC存在关联。其中,乳酸是糖酵解的最终产物,是肿瘤中常见的代谢产物,高水平的糖酵解代谢和乳酸积累也是肿瘤细胞的普遍特征之一。然而,代谢重编程(Metabolic reprogramming)如何影响表观遗传改变,特别是PDAC的组蛋白乳酸化,目前仍不清楚。

2024年5月6日,北京大学第三医院(北医三院)内分泌科魏蕊研究员/洪天配教授团队在Molecular Cancer以题目“Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma”发表了关于乳酸化修饰调控胰腺导管腺癌发生发展的研究成果。研究发现,与正常组织相比PDAC患者癌细胞中组蛋白乳酸化的水平显著升高,尤其是H3K18la水平较高,并且高水平的H3K18la与预后不良相关。抑制糖酵解活性或者敲低乳酸脱氢酶A(lactate dehydrogenase A,LDHA)能显著抑制H3K18la水平,并表现出显著的肿瘤抑制效果。H3K18la不仅能上调蛋白TTK和BUB1B的表达,上调后的 TTK和BUB1B还能进一步提高P300的表达,促进糖酵解和乳酸的积累,上调的乳酸水平又会进一步影响组蛋白的乳酸化。此外,TTK还可以促进LDHA的磷酸化(Y239),进而激活LDHA,进而上调H3K18la和乳酸水平,并进一步上调组蛋白的乳酸化水平。说明H3K18la-TTK/BUB1B-糖酵解/乳酸代谢之间存在正反馈调节通路促进PDAC的恶化。在本研究中为了明确乳酸代谢相关的基因和PDAC的关系,研究团队使用汉恒生物提供的sh-LDHA 和 OE-TTK慢病毒,成功构建了同时敲低HDLA和高表达TTK的MIA PaCa-2稳转细胞株。
下面,我们一起来了解具体的研究内容:
首先,研究团队发现PDAC患者胰腺组织中乳酸含量高于正常胰腺组织,对 MIA PaCa-2, PANC-1, AsPC-1 和 PL45等胰腺癌细胞系和胰腺导管上皮细胞系hTERT-HPNE的分析也得到了相同结果。对PDAC患者来源血清和健康对照血清进行非靶向代谢组分析,得到222个显著差异的代谢产物,其中122个上调,100个下调。KEGG分析发现,这些差异的代谢产物富集于中心碳代谢,其中包括乳酸。由于代谢产生的乳酸可以作为组蛋白乳酸化的底物,研究团队检测了PDAC组织和癌旁组织的组蛋白质乳酸化水平。在PDAC组织中观察到更高水平的泛赖氨酸乳酸化(Higher levels of global/pan-lysine lactylation,Pan Kla),这其中包括H3K18la。H3K18la被报道广泛参与多种生理生化过程,特别是肿瘤的发展,作者便聚焦于它的表达与PDAC的关系。通过病理分析和相关的医学统计发现H3K18la在PDAC组织中上调,并且AJCC等级越高H3K18la表达越高。
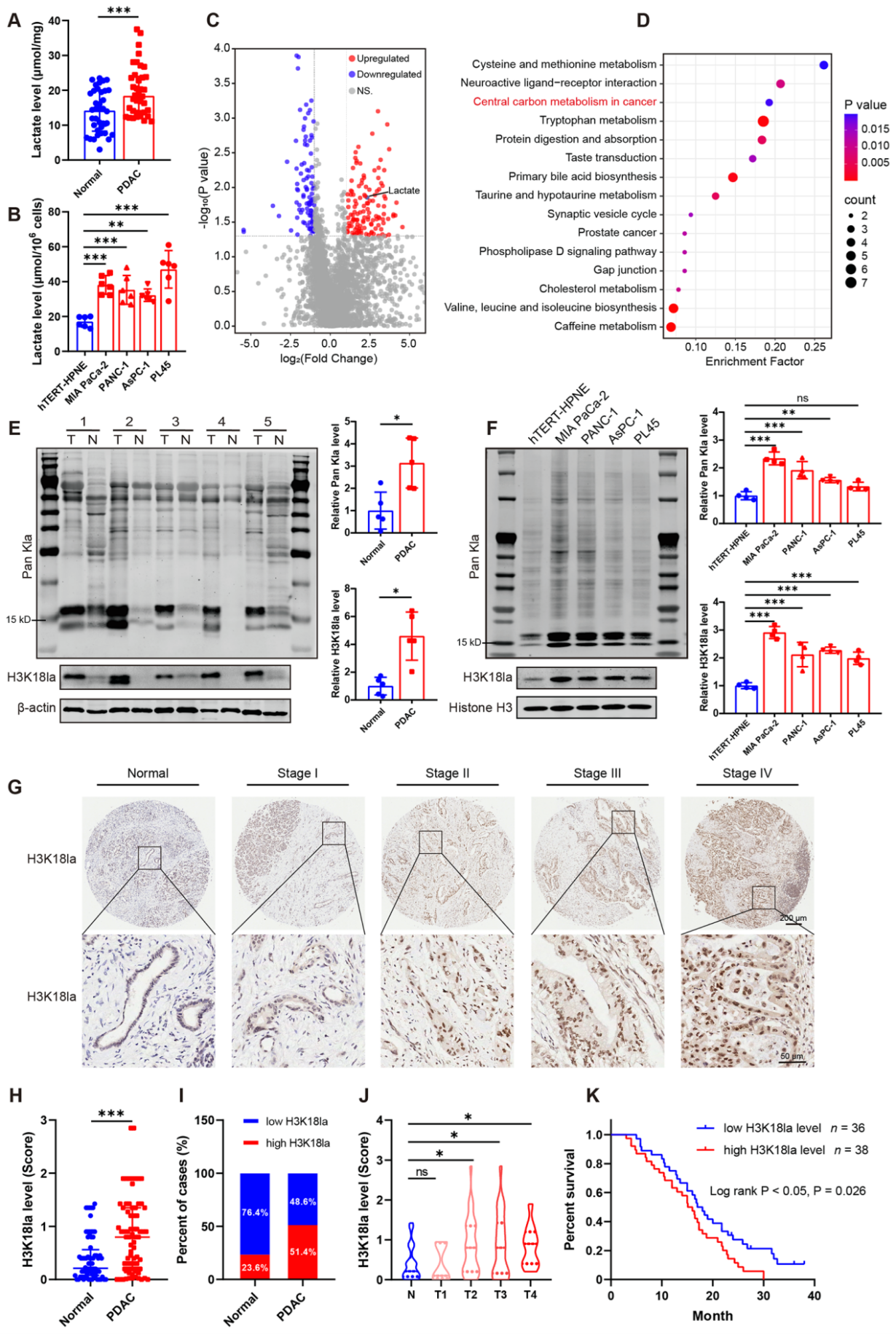
图1. 乳酸水平升高和组蛋白乳酸化与PDAC患者的不良预后相关
为了探索糖代谢升高是否和组蛋白的乳酸化存在关系,作者通过抑制剂抑制糖酵解的过程,或者直接在MIA PaCa-2和AsPC-1细胞中敲低LDHA。结果显示抑制剂能以剂量依赖形式显著下调Pan Kla 和 H3K18la水平,通过siRNA敲低LDHA的表达可以获得相似的效果。
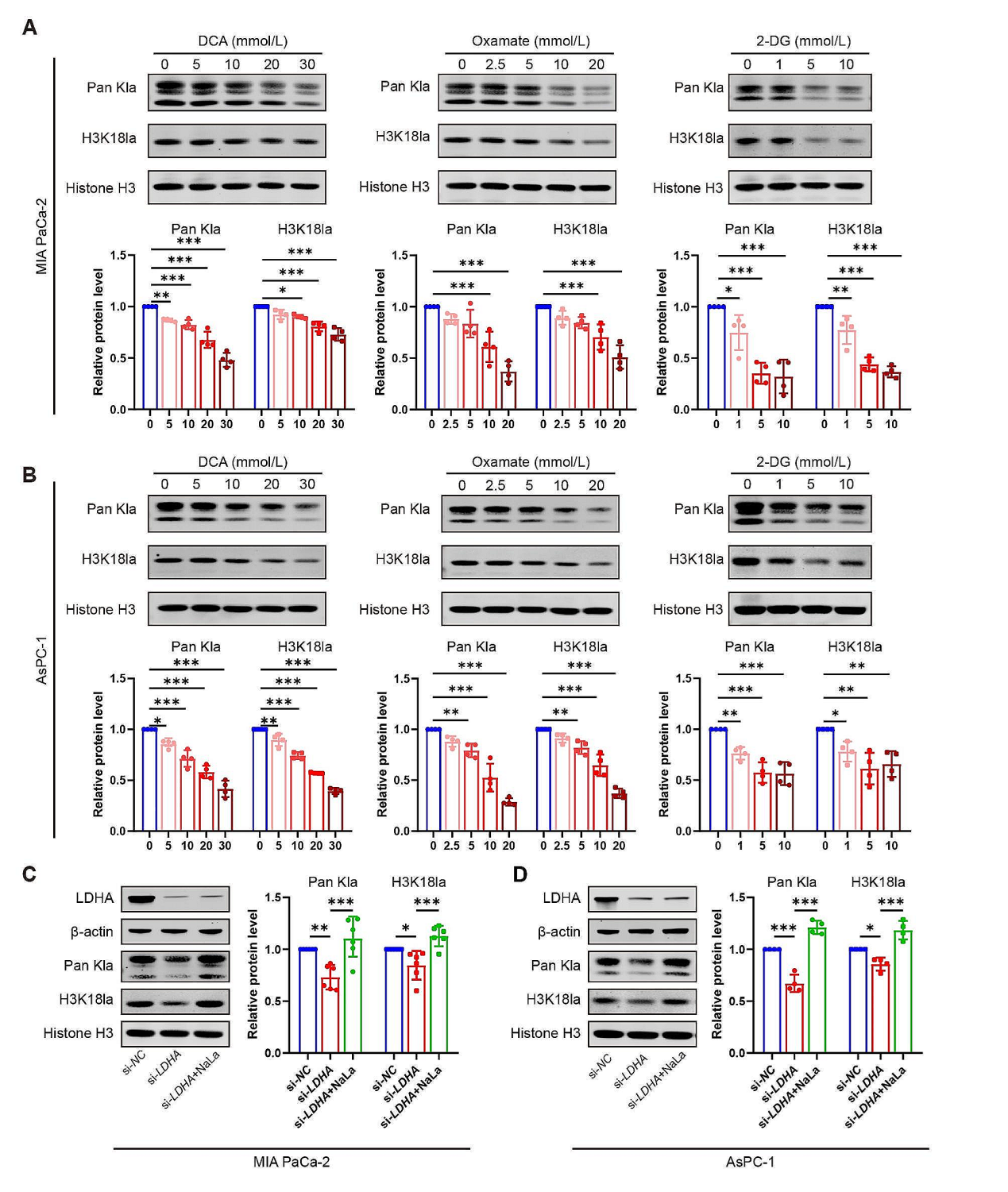
图2. 糖酵解抑制减少组蛋白乳酸化和抑制PDAC细胞增殖
随后,研究团队关注下调组蛋白乳酸化后对细胞功能的影响。首先使用不同抑制剂抑制细胞糖酵解,发现糖酵解被抑制后细胞生存能力和克隆形成能力显著减弱,siRNA敲低LDHA后细胞生长和克隆形成能力也显著降低。随后进行的划痕和细胞小室实验中,糖酵解抑制剂可以显著降低细胞的迁移侵袭能力,并且随抑制剂剂量升高而降低越明显。这些发现表明组蛋白乳酸化在PDAC的发生和发展中起着至关重要的作用,而抑制乳酸化可能具有潜在的抗肿瘤作用。
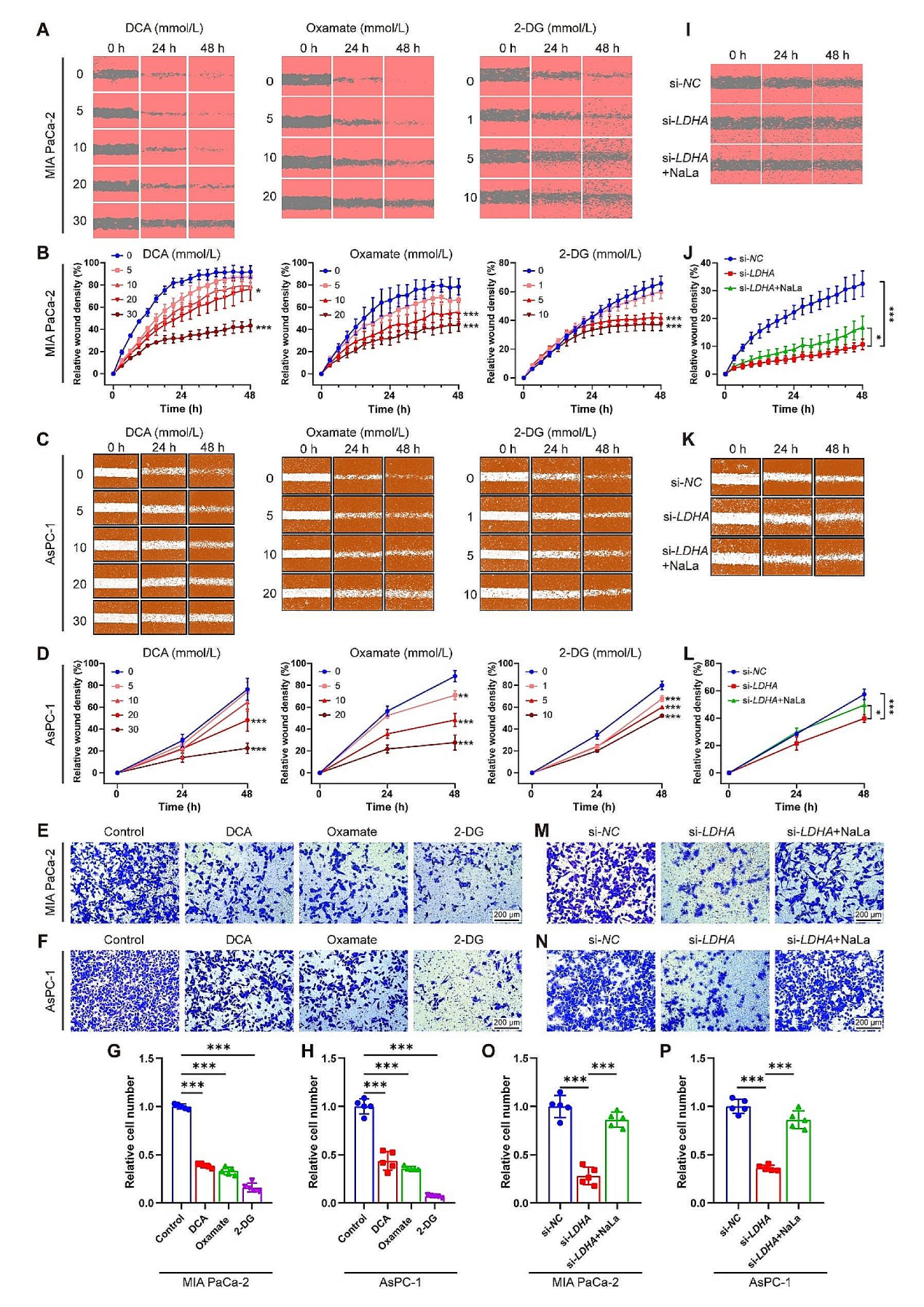
图3. 糖酵解抑制组蛋白乳酸化和PDAC细胞增殖和迁移
为了进一步研究组蛋白乳酸化的功能,研究团队将MIA PaCa-2细胞植入裸鼠皮下,随后分为两组分别腹腔注射糖酵解抑制(Oxamate)或PBS。与对照组相比,糖酵解抑制剂能显著下调H3K18la水平,并抑制肿瘤的生长。同样,为了研究LDHA敲低对肿瘤生长的影响,作者用sh-LDHA慢病毒构建了LDHA敲低的MIA PaCa-2 稳转株,将LDHA敲低MIA PaCa-2 稳转株植入到小鼠皮下,与糖酵解抑制剂处理相似,LDHA敲低组中的肿瘤体积和重量显著低于对照组。
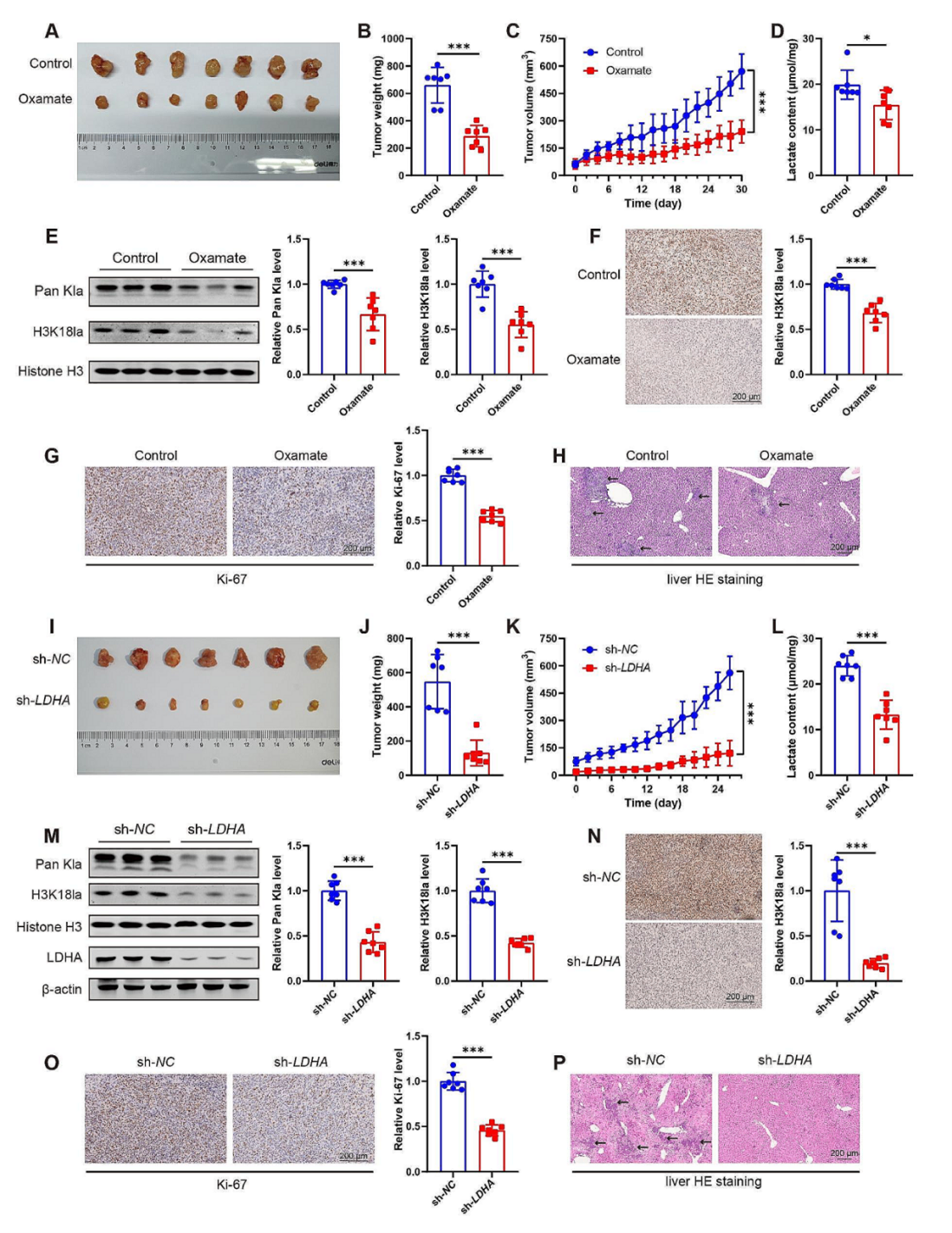
图4. 糖酵解抑制下调裸鼠肿瘤的组蛋白乳酸化并抑制癌症发展
证明PDAC细胞的乳酸代谢和组蛋白乳酸化存在关联后,研究团队展开分子机制的探索。P300已有报道参与组蛋白的乳酸化,但是目前没有关于P300和PDAC的相关研究。作者分别使用靶向P300的siRNA或者P300的抑制剂(C646)干预PDAC细胞系(MIA PaCa-2 和 AsPC-1),免疫印迹检测发现无论是用siRNA敲低P300或是抑制剂处理,H3K18la和Pan Kla水平都有所下调。在细胞功能实验上,siRNA或抑制剂抑制P300的功能后,细胞的增殖、迁移和侵袭能力有所下降。以上实验结果表明,P300很可能通过组蛋白的乳酸化参与PDAC的发展。
接下来作者尝试寻找下去组蛋白乳酸化相关的酶。通过抑制剂筛选发现,在众多参与组蛋白乳酸化的酶中,HDAC2的抑制剂能上调PDAC细胞组蛋白乳酸化水平,过表达HDAC2后能显著抑制PDAC细胞系组蛋白乳酸化,尤其是能下调H3K18la水平。以上结果表明P300和HDAC2分别参与了 PDAC细胞组蛋白乳酸化和去乳酸化。
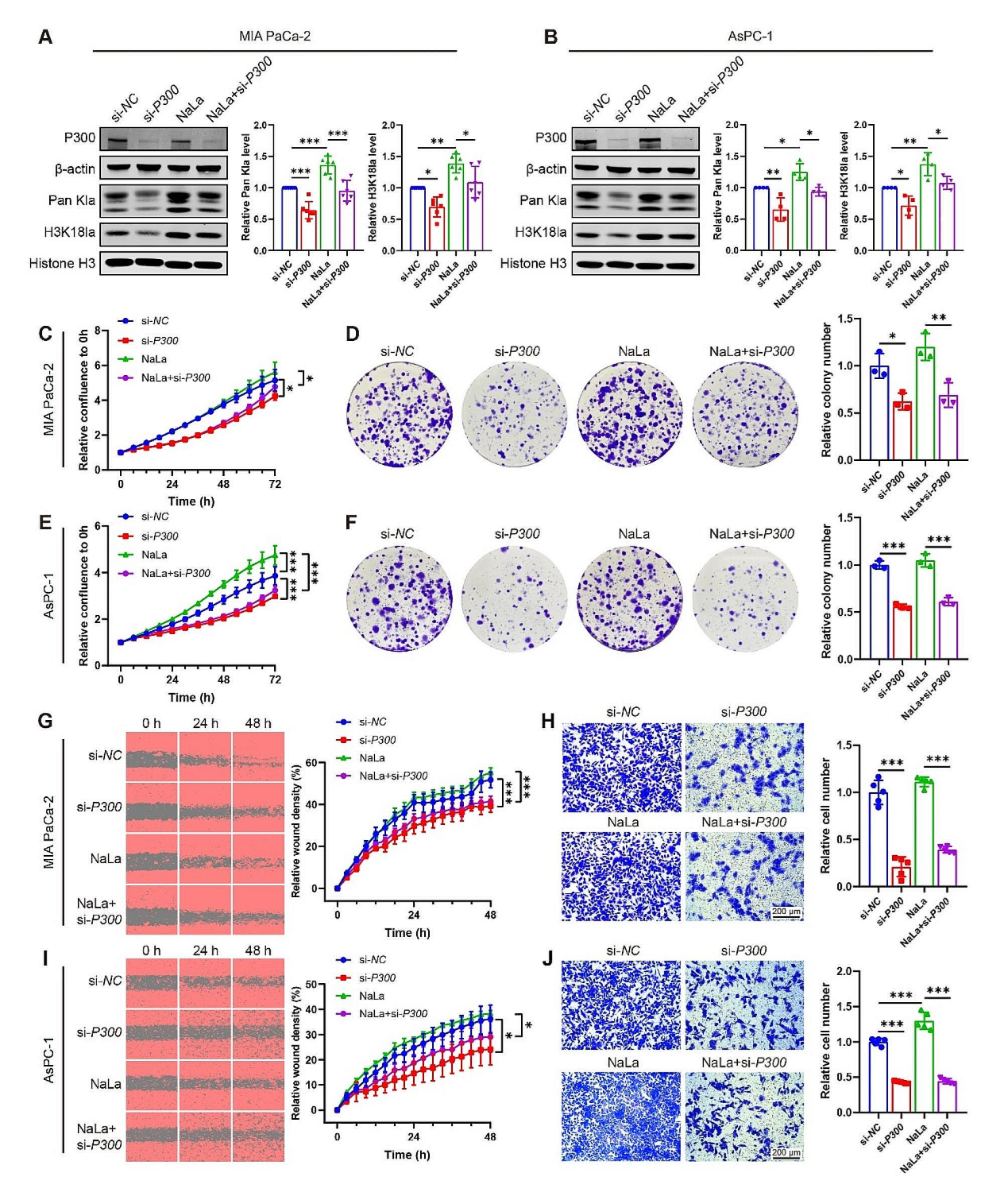
图5. P300和HDAC2参与了 PDAC细胞组蛋白乳酸化
随后,作者将尝试研究H3K18la如何影响PDAC疾病的发展。作者分别使用糖酵解抑制剂和PBS处理MIA PaCa-2系,并使用H3K18la抗体进行CUT&Tag分析,结果发现组蛋白乳酸化能影响细胞周期和肿瘤相关通路,如MAPK信号通路。同时通过RNAseq分析筛选受到乳酸化调控的靶基因,结果与cut-tag分析一致,大量基因富集于细胞周期相关通路中。综合分析GEPIA\CUT&TAG\RNAseq数据,发下14个在抑制剂处理细胞中下调并且在启动子区域富集H3K18la减少的基因,而在PDAC样本中趋势恰好相反。这14个基因中,CDC6, TTK, BUB1B, PTTG1与细胞周期相关,其中TTK, BUB1B在有丝分裂过程中至关重要,因此认为TTK和 BUB1B可能是组蛋白乳酸化调控的下游基因。ChIP-qPCR分析发现H3K18la在这两个基因的启动子处有明显的富集,糖酵解的抑制剂也能下调TTK 和 BUB1B 的表达。说明TTK 和 BUB1B的激活可能参与H3K18la调控的PDAC发展。
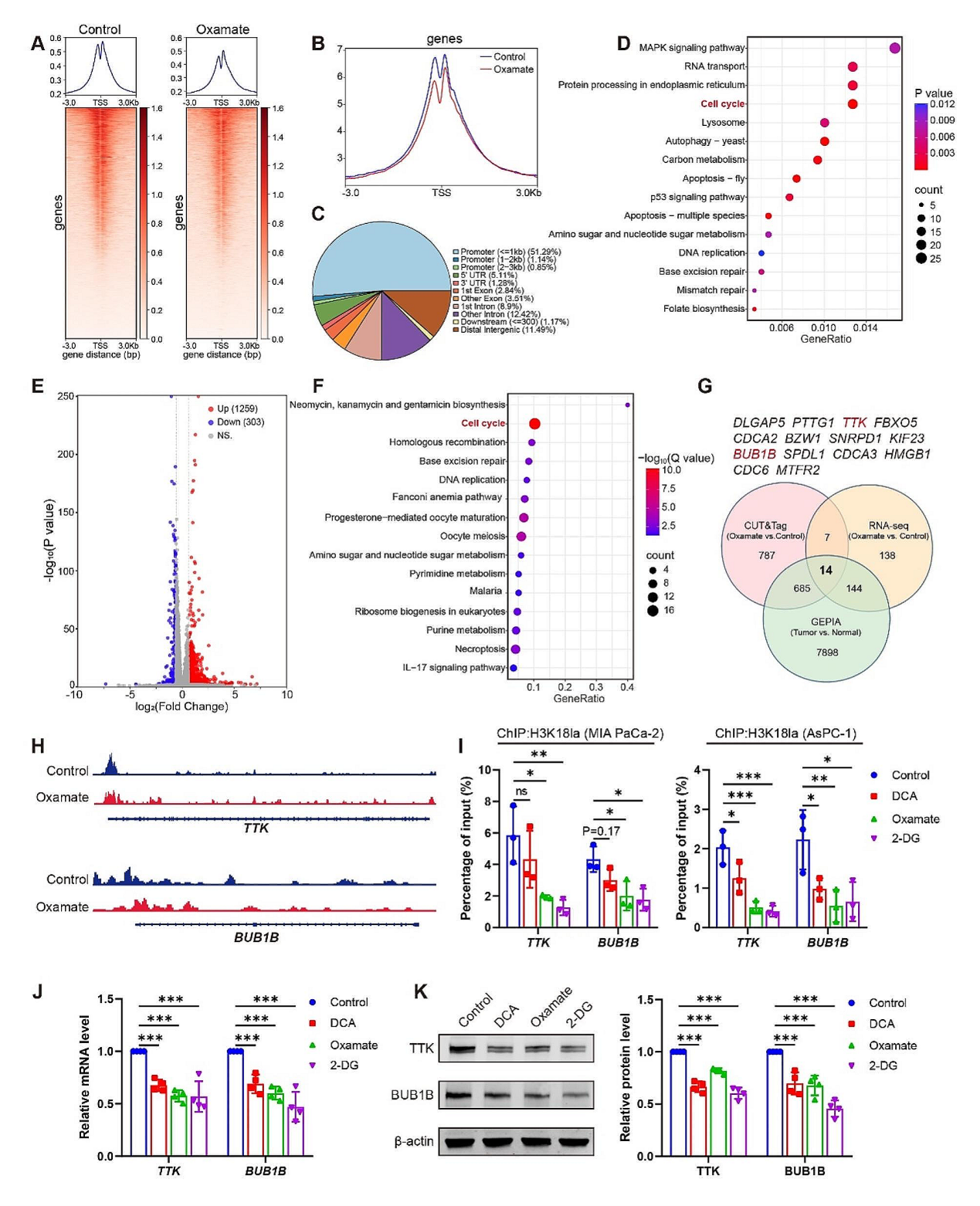
图6. H3K18la激活 TTK 和 BUB1B 的表达,进而影响PDAC的发展
为了探索TTK和BUB1B对PDAC发展的影响,首先检测了PDAC中这两个基因的表达水平。QPCR和WB实验检测发现,PDAC患者肿瘤组织的TTK 和 BUB1B显著高于癌旁组织,免疫组化分析也得到一致的结果。从GEPIA 和 Kaplan-Meier Plotter数据库获取的相关PDAC患者信息分析发现TTK 或 BUB1B水平高的患者总生存时间或无病生存时间较短。PDAC细胞系 (MIA PaCa-2, PANC-1, AsPC-1, and PL45 cells) TTK 和 BUB1B的表达水平也显著高于hTERT-HPNE细胞。相反,敲低TTK 或 BUB1B能显著抑制PDAC细胞系的增殖和迁移能力。这些研究结果表明TK和BUB1B参与了H3K18la调控的 PDAC恶性发展。
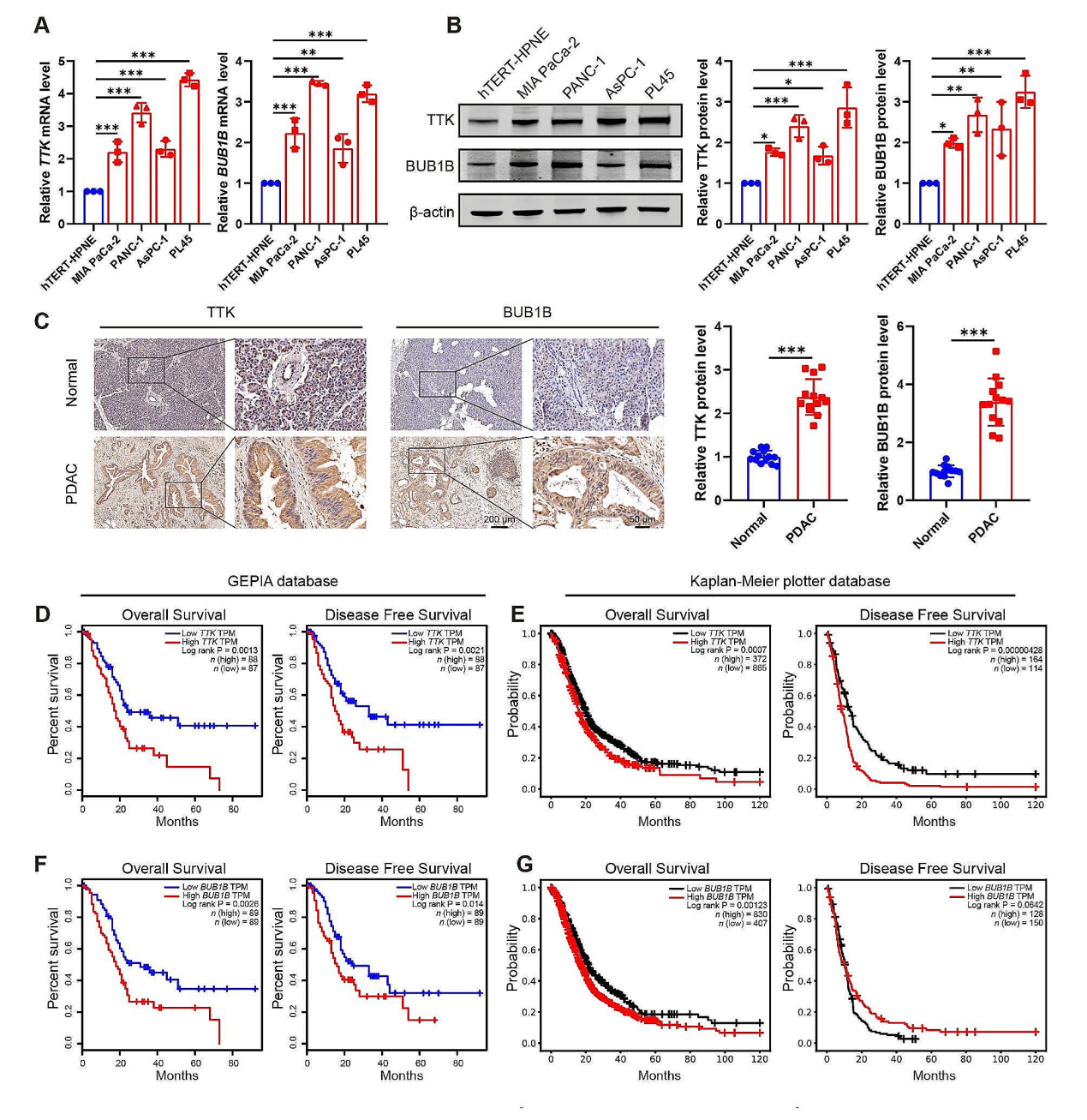
图7. TTK和BUB1B的高水平与PDAC的恶性相关
在以往的研究中BUB1B基因参与肺癌细胞的细胞周期与糖代谢的正向调控,敲低能下调糖酵解相关基因表达,包括solute carrier family 2 number 1、 LDHA、 pyruvate kinase M 2 和hexokinase2。因此,作者推测BUB1B能通过上调糖代谢的水平进而促进PDAC的生长。作者在研究中还发现敲低TTK和BUB1B能下调P300的水平,结合前文P300促进组蛋白乳酸化的发现,作者推测在PDAC发展过程中,除了组蛋白乳酸化—TTK/BUB1B通路,还有一个TTK/BUB1B整反馈调节乳酸化的机制。
随后作者研究了TTK/BUB1B是否通过影响乳酸化进而影响PDAC疾病的发展。LDHA是糖酵解代谢的关键酶之一,在糖酵解过程中,LDHA催化丙酮酸转化为乳酸。利用PhosphoSitePlus 工具,预测到LDHA存在两个磷酸化位点(Y10,Y239)。既然LDHA和乳酸产生密切相关,且含有磷酸化位点,其很有可能是TTK的作用靶点。CO-IP发现TTK会和LDHA存在相互作用,敲低TTK能显著抑制LDHA激活(磷酸化)水平,从而抑制乳酸的产生,同时也降低了Pan kla 和H3K18la水平。以上研究说明H3K18la靶基因TTK/BUB1B、糖酵解/乳酸代谢之间存在正反馈通路。
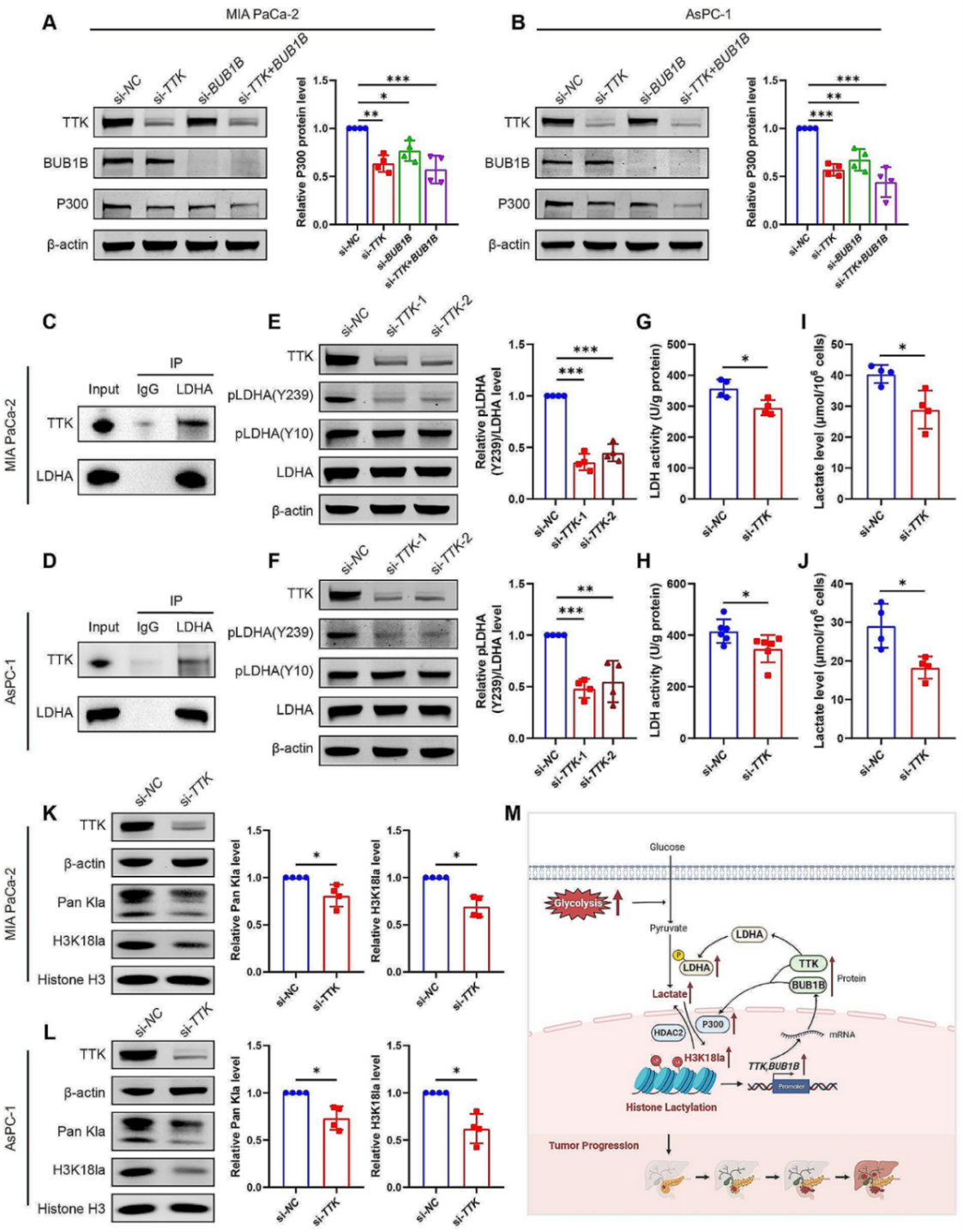
图8. H3K18la靶基因(TTK和BUB1B)与糖酵解之间存在正反馈通路
综合以上研究结果,乳酸促进肿瘤细胞增殖和迁移,其中一个重要因素就是组蛋白的乳酸化,尤其是H3K18la水平的升高,会上调TTK和BUB1B表达,TTK和BUB1B的上调又会进一步加剧糖酵解的发生,增加乳酸生成,形成一个正反馈通路,从而加剧肿瘤的恶化。
总而言之,本研究为代谢重编程和表观遗传调控提供了新的研究思路和补充。证实了H3K18la-TTK/BUB1B通路与能量代谢、表观遗传重编程和PDAC的关联,为攻克癌症提供了新的研究方向和治疗策略。









 8152
8152










